BICCN Pipelines
Overview
The Broad Institute’s Data Sciences Platform (DSP) develops production-level data processing pipelines in collaboration with multiple consortia including BICCN. Thank you to everyone who has worked with us to create and improve these pipelines. For more details please see our BICCN collaborators and how to cite the pipelines.
Pipelines are available for multiple data types hosted in the BICCN network, including single-cell and single-nucleus transcriptomics, methylomics, and ATAC-seq data.
Pipeline Standards, Maintenance, and Availability
Pipelines are cloud-optimized and developed to ensure portability as well as data reproducibility and interoperability. To this aim, the pipelines are:
- Open-access and developed with GA4GH standards.
- Written in the Workflow Description Language (WDL), a community-maintained, human-readable workflow language that can run on Cromwell, a portable execution engine that can be launched anywhere, locally or in the cloud.
- Containerized using public Docker instances, allowing researchers to exactly reproduce the workflow software.
The pipeline code is available from multiple sources, including GitHub, Dockstore, and the BCDC cloud computing environment (Terra).
- Code is developed and maintained in the WDL Analysis Research Pipelines (WARP) repository in GitHub. Overviews and workflow code for BICCN-related pipelines are linked in the table above; additionally, relevant pipelines can be identified by typing the keyword “BICCN” in the WARP Documentation search bar.The WARP Overview details navigating the repository, pipeline development, and running the workflows.
- Workflows are available for export from Dockstore, a GA4GH-compliant platform for sharing Docker-based tools. Search “warp” on Dockstore to find all WARP pipelines, including those used in the BICCN.
- Workflows are also available to test on Terra, the cloud-based bioinformatics platform used for BCDC data processing. To get started, register for Terra using the registration guide. To try a pipeline, navigate to the pipeline’s workspace linked in the table above or search for the “BICCN” tag in the workspaces tag search bar. Each workspace contains downsampled data, detailed instructions for using the workflows, and cost guidelines. Learn more about Terra with the Getting Started guides.
Citing the Pipelines
Each BICCN pipeline (see table above) has a SciCrunch resource identifier (RRID) that can be cited in publications. Follow the SciCrunch citation guidelines.
Example: (Optimus Pipeline, RRID:SCR_018908)
Additionally, please refer to the table above to cite any publications associated with the pipeline.
Additional Single-cell Transcriptomic Pipeline Resources
These pipelines produce outputs and quality control metrics that can be further analyzed and visualized with downstream community resources. Tutorials for combining single-cell transcriptomic data and pipelines with common community tools are available in the following resources:
BICCN Omics Workshop Workspace
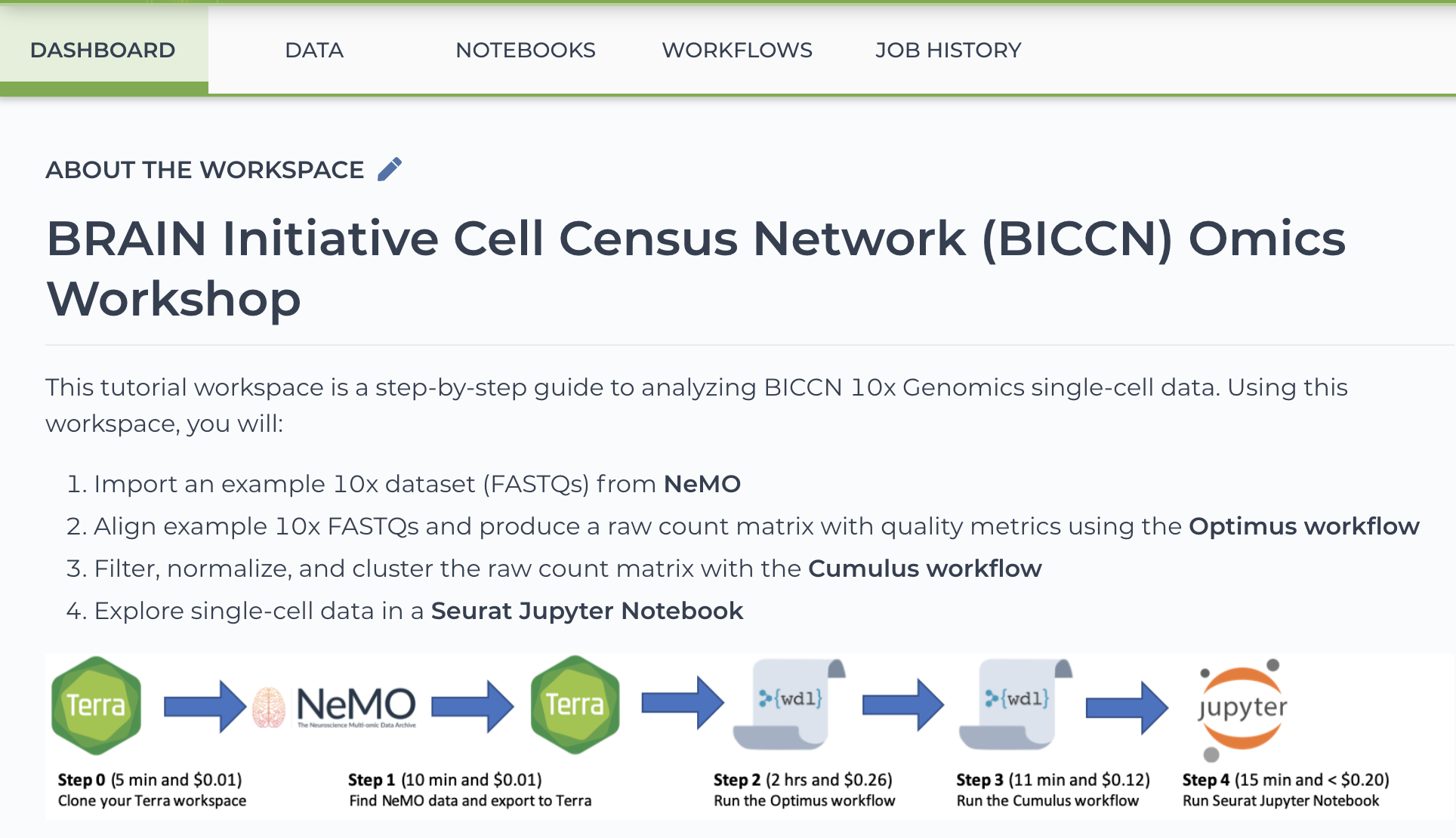
This tutorial Terra workspace is a step-by-step guide to analyzing BICCN 10x Genomics single-cell data. Using this workspace, researchers learn how to:
- Import an example 10x dataset (FASTQs) from NeMO
- Align example 10x FASTQs and produce a raw count matrix with quality metrics using the Optimus workflow
- Filter, normalize, and cluster the raw count matrix with the Cumulus workflow
- Explore single-cell data in a Seurat Jupyter Notebook
BICCN Omics Workshop Webinar Recording
This Brain Initiative Cell Census Network (BICCN) virtual workshop guides you through finding data in the Neuroscience Multi-Omic (NeMO) Archive, analyzing that data in Terra through workflows (pipelines) and interactive analysis, then publishing the results to a study in the Single Cell Portal (SCP).
BICCN Omics Workshop Blog
This high-level overview of the BICCN Omics Workshop describes the BICCN Omics workshop content and provides a link to the webinar demonstration.
Acknowledgments
We thank the following BICCN collaborators and Broad Pipelines Team members for their work on these pipelines:
MethylC-Seq (CEMBA)
Our gratitude to the Joseph Ecker Lab and special thanks to Joseph Ecker, Chongyuan Luo, Eran Mukamel, Hanqing Liu, Benjamin Carlin, Dan Moran, and Jeff Korte.
Single-Cell ATAC (scATAC)
Our gratitude to the Bing Ren Lab and special thanks to Bing Ren, Rongxin Fang, Yang Li, Sebastian Preissl, Nick Barkas, and Kylee Degatano.
Smart-seq2 Single Nucleus
Our gratitude to the Allen Institute, the Eran Mukamel Lab, and the NeMO team. Special thanks to Eran Mukamel, Fangming Xie, Zizhen Yao, Changkyu Lee, Jeff Goldy, Brian Herb, Cindy van Velthoven, Carrie McCracken, Kishori Konwar, Farzaneh Khajouei, Jessica Way, and Kylee Degatano.
Optimus
Our gratitude to Alex Dobin and the Eran Mukamel Lab. Special thanks to Kishori Konwar, Farzaneh Khajouei, Jessica Way, Ambrose Carr, Jishu Xu, Jose Soto, and Nick Barkas. This pipeline is currently being updated for the BICCN; more acknowledgments will be added as the work progresses.